
Hook
21 CFR Part 11 is one of the most cited rules in pharma quality discussions, and one of the easiest to oversimplify.
Teams often hear “Part 11” and think of software, passwords, or signatures. In practice, the rule is about something broader: whether electronic records and electronic signatures can be trusted the same way paper records are expected to be trusted.
For pharma teams, that matters because the record is not just a file. It is evidence.
Real-world scenario
Imagine a QA team that uses a password-protected spreadsheet for deviations, a shared folder for Corrective and Preventive Action (CAPA), and a separate tracker for training.
At first it feels workable. Then an auditor asks for the record history, approval trail, and final version of a change. The team can find the pieces, but only by rebuilding the story from several files.
That is the point where Part 11 becomes operational instead of theoretical.
What 21 CFR Part 11 means
21 CFR Part 11 is the FDA regulation that sets expectations for electronic records and electronic signatures in regulated environments.
At a practical level, it asks a simple question: can the system produce records that are secure, attributable, and reviewable over time“
That usually means the system needs to support:
- trustworthy electronic records
- meaningful electronic signatures
- audit trails where required
- record retention and retrieval
- controlled access and review
Why the rule matters
The FDA does not care whether the tool is fashionable. It cares whether the record is defensible.
That is why Part 11 matters across quality workflows such as:
- deviation management
- Corrective and Preventive Action (CAPA)
- document control
- training management
- change control
If the process is electronic, but the evidence still depends on manual stitching, the organization has not really escaped paper habits. It has only moved them into a browser window.
Why the FDA requires it
The purpose of Part 11 is to prevent electronic records from becoming easy to alter, hard to trace, or impossible to reconstruct.
The expectation is not perfection. The expectation is control:
- know who did what
- know when it happened
- know why it changed
- preserve the history
- keep the final record trustworthy
Comparison at a glance
- Validation: The system is fit for intended use
- Audit trail: Changes can be traced without losing history
- Electronic signature: The approval is tied to a person and an action
- Record retention: The evidence survives over time
- Access control: Only the right people can modify or approve records
This is where the discussion becomes practical. The regulation is not asking for ceremony. It is asking for proof.
What teams usually get wrong
Most Part 11 problems do not start with bad intent. They start with weak workflow design and too much trust in manual controls.
- Common assumption: A password makes a record compliant
What really happens: A password only controls access; it does not prove record integrity - Common assumption: A timestamp proves control
What really happens: A timestamp is useful, but it does not replace traceable approvals - Common assumption: A spreadsheet can be “validated” easily
What really happens: Once spreadsheets support regulated records, validation and change control become much harder - Common assumption: Electronic signatures are just digital initials
What really happens: Part 11 signatures have to be linked to the action and governed properly
Where Part 11 applies
Part 11 becomes relevant when electronic systems are used to create, maintain, approve, or store regulated records.
That often includes:
- electronic deviation records
- Corrective and Preventive Action (CAPA) approvals
- Standard Operating Procedure (SOP) updates in document control
- training completion records
- change control approvals
In other words, the rule shows up wherever a quality team needs to prove that a digital record is still a reliable record.
What the FDA guidance actually emphasizes
FDA’s Part 11 guidance is useful because it points teams toward the controls that matter most in practice.
The focus is on areas such as:
- validation
- audit trails
- record copies
- retention
- legacy systems
That gives teams a practical way to review readiness without turning the topic into legal theory. The takeaway is not “buy software.” The takeaway is “design the system so the record is defensible.”
What inspectors are really testing
Inspectors are not just checking whether a tool exists. They are checking whether the record can still be trusted when the process is reviewed later.
That means they care about whether:
- the action is attributable
- the signature is meaningful
- the history is traceable
- the final record can be retrieved and understood
If the answer depends on someone rebuilding the story from multiple files, the process is weaker than it looks.
Callout:
the real test Part 11 is not asking whether the team has software. It is asking whether the evidence still holds together when an inspector asks to see the full story.
Audit trail, validation, and retention
The strongest Part 11 discussions usually come back to four control points:
- Validation: the system has to be fit for intended use
- Audit trail: changes should not obscure earlier entries
- Copies of records: investigators need reasonable access to useful copies
- Record retention: content and meaning have to survive over time
Those are exactly the areas where spreadsheet-heavy processes become fragile.
What a better system changes
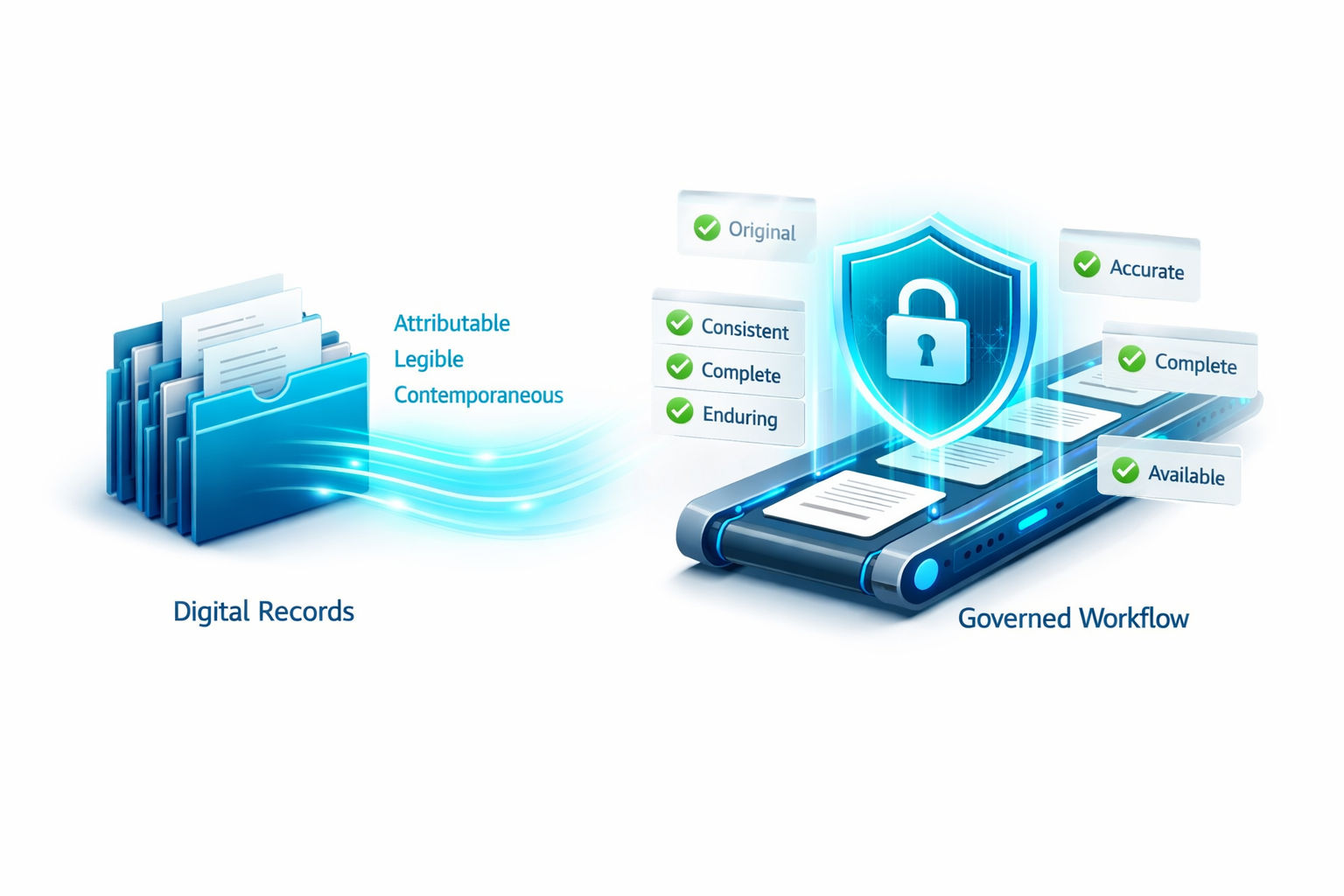
A controlled eQMS does not just move the data into a new place. It changes the way the evidence is formed.
The action happens inside a governed workflow. Approvals are attached to the record. The history stays with the record. Review is visible instead of reconstructed later.
That is what makes Part 11 easier to defend without making the process heavy.
Download template
If your team is reviewing Part 11 readiness, a short checklist is the most useful next step.
The checklist should help teams ask:
- Which records are electronic“
- Where do signatures attach“
- Can changes be traced cleanly“
- Can records be copied and retained without losing meaning“
That is a practical pre-assessment before a broader system review.
Complere fit
Complere is relevant here because Part 11 is really a workflow and evidence problem, not just a file format problem.
The platform becomes useful when teams need to keep records governed, approvals visible, and the compliance story attached to the action instead of built afterward.
Closing thought
Part 11 is easier to explain than to demonstrate.
If the team can show the record, the signature, the history, and the rationale without rebuilding the story from scattered files, they are in a far stronger position.
Disclaimer
This article is a practical interpretation of the FDA’s Part 11 guidance and is not legal advice. Teams should confirm specific requirements against their predicate rules, intended use, and internal validation approach.

.jpeg)
.jpeg)
.jpeg)
